
Treatments
Current Treatment Approaches for Duchenne Muscular Dystrophy
Duchenne muscular dystrophy (DMD) is a thief of motion, a genetic specter passed silently along the X chromosome, where a single flawed gene unravels the body’s ability to craft dystrophin—that quiet sentinel tasked with shielding muscle fibers from their own exertion (1). Picture it: dystrophin, a scaffold of resilience, weaving itself into a greater tapestry of proteins—the DAPC—to steady the shuddering dance of contraction. Without it, muscles fray like a rope under strain. In those born with DMD, this absence is a slow-burning fuse. Fibers split, inflammation smolders, and scar tissue creeps in like kudzu, strangling what little chance remains for repair (3). The body, ever faithful to its broken blueprint, becomes a prisoner of its own architecture—a tragedy written in every faltering step.

In the early years, when life should hum with the untamed energy of discovery, Duchenne muscular dystrophy begins its quiet siege. Picture a child, three summers old, whose legs falter where others sprint—a delay in milestones as subtle as a shadow lengthening at dusk. By eight or ten, the body’s scaffolding buckles, braces, and wheelchairs become lifelines, steel, and plastic against the slow unraveling of muscle. A cruel arithmetic follows: breaths shallowed, heartbeats frayed, until the body surrenders to its own exhausted biology (4). This is not merely a clinical narrative but a ledger of loss—emotional and financial, a weight shouldered by families and hospitals alike, etched into spreadsheets and sleepless nights (5).
Now, shift the lens. Zoom past the visible to the molecular theater, where DNA’s alphabet spells tragedy or reprieve. Here, mutations are sentences mangled by missing words. An “out-of-frame” deletion is a paragraph gutted, syntax scrambled into nonsense—a dystrophin protein truncated, useless, leaving muscles defenseless as unmoored sails. This is DMD: a body adrift. Here, dystrophin stumbles but staggers on, a compromised sentinel. This is Becker’s, a slower unraveling (6).
Imagine the sarcolemma as a fortress wall. Without dystrophin’s glue, the bricks loosen. Calcium storms breach the gates, sparking chaos—a cascade of inflammation, necrosis spreading like wildfire, fibrosis paving muscle with scar. Each step is a domino fall, a clockwork of collapse (7). The body, in the end, is a story of structure. And the structure is everything: from the arc of life to the dance of atoms that sustain it.
Over the past few decades, multiple DMD animal models have been developed to understand the pathogenesis and evaluate therapeutic strategies. The mdx mouse is the most commonly used model, bearing a point mutation in exon 23 of the murine DMD gene (8). Although mdx mice mirror many pathological features of DMD in humans, they exhibit a milder phenotype partly due to compensatory mechanisms by utrophin and α7-integrin (9). Other more severe models (e.g., utrophin-dystrophin double-knockout mice) (10) and DMD dog models (11) also exist, offering specific advantages for translational research.
To date, there is still no definitive cure for DMD. Standard therapy typically involves managing symptoms and delaying disease progression through glucocorticoid therapy (12). However, multiple lines of research are focused on two broad therapeutic approaches:
-
- Dystrophin-targeted therapies, including gene-based, cell-based, and protein replacement strategies, aim to restore partial or full dystrophin function.
- Downstream pathophysiology-modulating strategies targeting inflammation, fibrosis, oxidative stress, impaired calcium homeostasis, muscle ischemia, and muscle atrophy to mitigate disease severity.
This comprehensive review will present current and emerging therapeutic strategies in both categories, focusing on those that have reached clinical investigation or exhibit strong potential for clinical translation. Furthermore, we will present a meta-analysis of gene profiling data obtained from patients with DMD to illustrate key molecular mechanisms and novel candidate gene targets that may inform future therapies.

Therapeutic Strategies Targeting Dystrophin
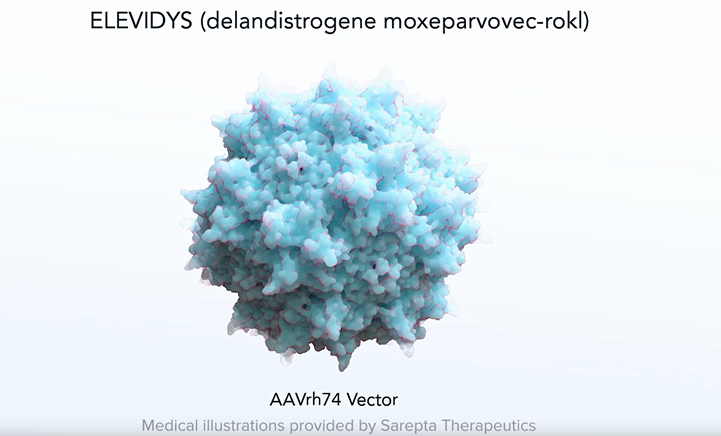
Currently, the most promising treatment for targeting dystrophin is ELEVIDYS
ELEVIDYS is an adeno-associated virus (AAV) vector–based gene therapy intended to deliver a shortened but functional version of the dystrophin gene—often called a “micro-dystrophin”—into muscle cells. This micro-dystrophin aims to compensate for the defective or missing dystrophin that characterizes DMD. By restoring partial dystrophin function, ELEVIDYS may help stabilize muscle fibers, reduce damage, and potentially improve muscle strength and function.
In June 2023, the U.S. Food and Drug Administration (FDA) granted accelerated approval for ELEVIDYS to treat ambulatory pediatric patients aged 4 through 5 years with a confirmed mutation in the dystrophin gene (Food and Drug Administration, 2023, https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-gene-therapy-muscular-dystrophy). This approval marked the first gene therapy for muscular dystrophy and represented a significant milestone in the treatment of this rare disorder. Since the FDA’s accelerated approval of Elevidys in June 2023, no major expansions to its indicated age range have been officially approved as of early 2025. Sarepta Therapeutics continues confirmatory trials to evaluate Elevidys in broader age groups and to gather more long-term safety and efficacy data.
Benefits of ELEVIDYS
-
- Targeting the Genetic Root Cause
By delivering a functional version of the dystrophin gene to muscle cells, ELEVIDYS addresses the primary defect responsible for DMD. This contrasts with symptomatic treatments that primarily manage complications without correcting the underlying pathology. - Potential for Slowing Disease Progression
Early clinical data suggest that ELEVIDYS may reduce muscle fiber damage and slow the rate of decline in muscle function. While not a cure, the therapy’s intent is to help preserve muscle strength and prolong ambulation. - Reduced Need for Some Supportive Therapies
If ELEVIDYS effectively stabilizes muscle function over the long term, it could reduce reliance on high-dose steroids. This is noteworthy given the side effects associated with prolonged steroid use, such as bone density loss and weight gain. - Single-Dose Administration
As a one-time gene therapy, ELEVIDYS eliminates the need for repeated deliveries of micro-dystrophin. This can be advantageous compared to therapies that require continuous administration.
- Targeting the Genetic Root Cause
Limitations and Challenges
-
- Age and Ambulatory Status Restriction
Current FDA accelerated approval is limited to ambulatory pediatric patients aged 4 through 5. Patients outside this age range or those who are no longer ambulatory are still under investigation or may not currently qualify for ELEVIDYS. - Uncertain Long-Term Efficacy
While short-term data are promising, the long-term durability of micro-dystrophin expression and clinical benefit remain under investigation. The FDA’s accelerated approval requires confirmatory trials to demonstrate sustained safety and efficacy. - Potential Immune Responses
Gene therapy using an AAV vector can trigger immune responses. Patients can develop antibodies against the AAV capsid, which could limit the therapy’s effectiveness and complicate potential future treatments. Closely monitoring liver function and overall immune response is part of post-treatment care. - High Cost and Limited Availability
Like many gene therapies, ELEVIDYS is associated with a significant cost, and the degree of insurance coverage can vary. Limited manufacturing capacity and the complexity of gene therapy production can also affect availability. - No Complete Cure
Despite its potential, ELEVIDYS is not a cure. Restoring full dystrophin production remains an elusive goal, and this therapy offers a truncated dystrophin. Symptomatic management and supportive care continue to be essential.
- Age and Ambulatory Status Restriction
Moving Forward
The development of ELEVIDYS underscores the remarkable progress made in gene therapy. Although it has limitations—especially regarding long-term data, patient selection, cost, and potential immune responses—this therapy provides a new path to address the core genetic defect in DMD. Ongoing clinical trials will clarify its long-term benefits and risks in broader patient populations.
For families and patients considering ELEVIDYS, discussions with a multidisciplinary care team—comprising neurologists, geneticists, and rehabilitation specialists—are essential to determine eligibility, manage potential side effects, and integrate gene therapy into a comprehensive treatment plan.
Other approaches to dealing with dystrophin absence
Since DMD is fundamentally caused by a functional dystrophin protein’s absence (or near absence), another attractive goal has been to correct or bypass the genetic defect. Several technologies aim to restore dystrophin expression:
-
- Gene-based therapies (e.g., exon skipping, stop codon readthrough, gene-addition strategies, gene editing).
- Cell-based therapies use transplanted cells that carry functional copies of the dystrophin gene.
- Protein replacement therapies (often leveraging utrophin or other proteins as substitutes or supplements for lost dystrophin function).
Gene-Based Therapeutic Strategies
Exon Skipping
Exon skipping involves using antisense oligonucleotides (AONs) designed to modulate pre-mRNA splicing. By forcing muscle cells to skip mutation-containing exons (e.g., exon 51, exon 53), the reading frame can be restored in certain DMD genotypes, leading to a shorter yet partially functional dystrophin (13).
-
- Eteplirsen (Exondys 51) became the first FDA-approved exon-skipping therapy in 2016, specifically targeting exon 51. It has shown promise in improving the 6-minute walk test (6MWT) distance in a subset of patients (14).
- Golodirsen and Viltolarsen—approved by the FDA in 2019 and 2020, respectively—target exon 53. Clinical trials demonstrated that both drugs increased dystrophin protein expression after 40–80 weeks of treatment, though the extent of functional improvement and long-term benefit is still under investigation (15).
The main limitation of exon-skipping therapy is that each medication typically only benefits patients with specific mutations amenable to a given exon skip (e.g., ∼14% for exon 51 skipping). Additionally, long-term efficacy and safety data remain sparse, and mild to moderate hypersensitivity reactions have been reported (16). Efforts are ongoing to develop multi-exon skipping therapies (e.g., double exon skipping), which could theoretically broaden the treated population, but optimizing in vivo delivery and splicing correction efficiency has proved challenging (17).
Stop Codon Readthrough
Around 10–15% of DMD cases stem from nonsense mutations producing premature stop codons, resulting in truncated dystrophin (18). Small molecules like Ataluren (formerly PTC124) enable the ribosome to read through these premature termination signals, potentially restoring some dystrophin production.
-
- Gentamicin, an aminoglycoside antibiotic, can enable readthrough but poses significant risks of renal toxicity and ototoxicity (19).
- Ataluren has shown efficacy in preclinical studies; the European Medicines Agency (EMA) granted it conditional approval for DMD patients with nonsense mutations (20). However, subsequent Phase 3 trials in the U.S. failed to show a significant improvement in motor function, and the FDA has not approved it (21).
Gene Addition
Since the DMD gene is unusually large, direct replacement with the full-length gene exceeds the capacity of conventional adeno-associated virus (AAV) vectors. Researchers have instead developed shorter but partially functional transgenes known as micro-dystrophin (22).
-
- Specific AAV serotypes (e.g., AAV9, AAV8, AAVrh74) offer enhanced muscle tropism.
- Clinical trials using PF-06939926 (micro-dystrophin transgene in an AAV9 vector) show increased dystrophin expression in muscle and improved motor function (23).
- Another approach uses rAAVrh74.MHCK7.micro-dystrophin, which has demonstrated robust muscle delivery in mice (24).
Challenges remain regarding the high doses of AAV required to target all skeletal muscles and the heart, leading to immunotoxicity and other safety concerns (e.g., inflammatory responses to the vector) (25).
Gene Editing
The CRISPR/Cas9 gene-editing system has recently revolutionized molecular biology, showing the potential to correct the underlying DMD gene defect. CRISPR/Cas9 can delete or repair out-of-frame mutations by introducing double-stranded breaks near mutated exons, effectively shifting them to an in-frame reading (26). In mdx mice, CRISPR/Cas9 has partially restored functional dystrophin and improved muscle strength (27).

However, significant hurdles persist:
-
- Immunogenicity to the Cas9 enzyme or the AAV vector.
- Off-target effects—undesirable edits elsewhere in the genome with potentially harmful consequences (28).
- Efficiency of delivery, requiring large vector doses.
Despite these complexities, gene editing remains an exciting, potentially curative avenue, especially once more refined CRISPR variants (e.g., base editors, prime editors) demonstrate safe and reliable in vivo efficacy (29).
Cell-Based Therapeutic Strategies
Cell-based therapies aim to introduce normal genomes and robust regenerative cells into dystrophic muscles. A key requirement is that these cells differentiate into myofibers and ideally replenish muscle stem cells (satellite cells) for sustainable repair (30).
-
- Satellite cell transplantation has shown promise in mdx mice but limited success in human trials, partly due to poor engraftment and immune rejection (31).
- CD133+ cells, derived from normal human skeletal muscle, can generate dystrophin-positive myofibers in mdx mice (32). However, clinical benefit remains ambiguous.
- Human-induced pluripotent stem cells (hiPSCs) from DMD patients can be genetically corrected (e.g., via CRISPR/Cas9) and then differentiated into myogenic progenitors. Early studies show restored dystrophin expression in mdx mice (33).
Major obstacles to clinical translation include:
-
- Low survival and limited migration of transplanted cells in diseased muscle.
-
- Immunogenicity and the long-term safety of hiPSC-derived products.
Hence, better scaffold technologies, immunosuppressive regimens, and improved reprogramming methods could boost engraftment, offering a cell-based replacement or adjunct to gene therapy (34).
Protein Replacement
Because restoring full-length dystrophin is so challenging, an alternative strategy is to upregulate or supplement dystrophin with functionally similar proteins.
-
- Utrophin, a structural and functional analog of dystrophin, is highly expressed during fetal development and in regenerating muscle. Overexpression of utrophin can protect mdx mice from severe dystrophic pathology (35).
-
- The small molecule Ezutromid (C1100) was designed to increase utrophin expression. However, clinical trials showed minimal efficacy, leading to termination of development (36).
-
- Another approach is GALGT2 gene transfer using an rAAVrh74 vector, aiming to up-regulate utrophin and reduce muscle damage (37).
While utrophin-based therapies may partially compensate for dystrophin absence, they do not completely replicate dystrophin function, such as the organization of sub-sarcolemmal microtubules (38). Combinational regimens integrating utrophin upregulation with gene or cell therapy approaches may offer more robust benefits.
Therapeutic Strategies Targeting Secondary Downstream Pathological Mechanisms
In addition to direct dystrophin restoration, there is a parallel focus on mitigating the deleterious downstream consequences of dystrophin loss. Key pathophysiological pathways include:
-
- Fibrosis – Excess extracellular matrix (ECM) deposition replacing functional muscle.
- Chronic inflammation – NF-κB, TNF-α, and other cytokine-mediated pathways.
- Ca^2+ dysregulation – Mechanosensitive channel leakage, mitochondrial dysfunction.
- Oxidative stress – Elevated reactive oxygen species (ROS) damaging proteins and lipids.
- Muscle ischemia – Decreased nitric oxide synthase activity, impaired vasodilation.
- Muscle atrophy – Myostatin, proteolytic pathways.
- Bone loss – Glucocorticoid-induced osteoporosis, reduced mechanical load, and vitamin D deficiency.
Many interventions target these disease modifiers to preserve muscle function, slow disease progression, and enhance quality of life, regardless of mutation type.
Therapeutic Strategies Targeting Fibrosis
In DMD, progressive fibrosis gradually replaces necrotic/regenerating muscle fibers. Muscle fibrosis correlates strongly with disease severity and functional decline. The key fibrogenic mediators include:
-
- Transforming Growth Factor-β (TGF-β)
- Connective Tissue Growth Factor (CTGF)
- Tumor Necrosis Factor-α (TNF-α)
Excessive TGF-β activation is considered a primary driver of ECM accumulation, fibroblast proliferation, and inflammation.
TGF-β Inhibition
-
- Losartan, an angiotensin II type 1 receptor blocker, can attenuate TGF-β signaling. Long-term Losartan partially alleviates diaphragm fibrosis in mdx mice, though human trials are inconsistent regarding functional benefits.
- Halofuginone (Halo) blocks TGF-β-dependent Smad3 phosphorylation, reducing myofiber fibrosis in mdx mice. However, at higher doses, Phase 1/2 trials were terminated after a serious adverse event.
- Suramin downregulates TGF-β signaling, lowering creatine kinase levels and fibrosis in mdx muscle. Preclinical data are promising, but more clinical data are needed.
- Imatinib mesylate (Gleevec®) also inhibits TGF-β-driven pro-fibrotic events. In certain models, it improved hindlimb grip strength and reduced fibrosis, yet other mdx strains showed no benefit or adverse cardiac effects.
CTGF Inhibition
CTGF is a key downstream mediator of TGF-β and strongly associated with fibroblast proliferation. It is notably elevated in mdx mice and DMD patients.
-
- FG-3019 is a fully human monoclonal antibody targeting the CTGF VWC (von Willebrand factor type C) domain, potentially interrupting fibrotic signaling. A Phase 2 trial in non-ambulatory DMD patients (NCT01890265) is underway. CTGF inhibition may be safer than direct TGF-β inhibition, given TGF-β’s broader physiological roles.
Therapeutic Strategies Targeting Inflammation
Dystrophic muscle is characterized by continual necrosis-regeneration cycles that trigger persistent inflammation. Elevated levels of Ca^2+ can activate the NF-κB pathway, leading to infiltration of macrophages, neutrophils, and other inflammatory cells.
Glucocorticoids remain the gold standard for anti-inflammatory therapy in DMD, prolonging ambulation by a few years, improving respiratory function, and delaying cardiomyopathy. Common glucocorticoids include prednisone, prednisolone, and deflazacort. However, side effects (e.g., weight gain, osteoporosis, metabolic disturbances) limit their long-term use.
New-generation glucocorticoid analogs aim to reduce adverse outcomes:
-
- Vamorolone is engineered to retain anti-inflammatory effects with fewer metabolic and endocrine side effects. Early-phase trials have shown promising safety and tolerability.
Other anti-inflammatory approaches:
-
- Flavocoxid (a dual COX-2/5-LOX inhibitor) reduced serum interleukin-1β and TNF-α levels in DMD patients, though short-term trials showed no significant functional improvement.
- TNF-α inhibitors such as Remicade (Infliximab) partially improved muscle strength and reduced inflammation in mdx mice, but potential cardiac side effects remain a concern.
- Tamoxifen (TAM), a selective estrogen receptor modulator, has shown beneficial effects on muscle force in mdx mice by modulating NF-κB-related inflammation and calcium homeostasis. A Phase 3 clinical trial (NCT03354039) is ongoing, although the exact mechanism is not fully clarified.
- Histone deacetylase (HDAC) inhibition with Givinostat downregulates inflammatory genes and fibrotic regulators in mdx mice. A Phase 3 trial (NCT02851797) is in progress, with earlier Phase 2 data showing increased muscle fiber size but uncertain functional benefits.
- Edasalonexent inhibits NF-κB to preserve muscle mass, reduce fibrosis, and improve endurance in mdx mice. Early human trials indicate a generally favorable safety profile, although mild gastrointestinal side effects occur.
- Recombinant human insulin-like growth factor-1 (rhIGF-1, Increlex) modifies inflammation and aids muscle regeneration. However, clinical trials have shown improvements in linear growth but negligible motor gains in DMD.
- TAS-205, an inhibitor of hematopoietic prostaglandin D synthase (HPGDS), may reduce levels of the proinflammatory mediator prostaglandin D2. Phase 1/2 data indicate safety and hints of efficacy, but larger trials are needed.
Therapeutic Strategies Targeting Muscle Damage
Ca^2+ Dysregulation
One hallmark of dystrophic muscle is excessive sarcolemmal fragility, enabling abnormal Ca^2+ influx. Cytosolic Ca^2+ overload triggers proteolytic enzymes (e.g., calpains), perpetuating muscle damage.
-
- Streptomycin is a broad-spectrum antibiotic that nonspecifically blocks stretch-activated ion channels, reducing muscle injury in mdx mice. However, it provides minimal benefits to the diaphragm and heart.
-
- GsMTx4 (AT-300), a specific mechanosensitive channel blocker, has improved muscle force in mdx mice. It received orphan drug designation but requires further study.
-
- Recombinant human MG53 augments membrane repair capacity, preventing fiber necrosis after muscle injury in mdx mice.
Rimeporide, an inhibitor of the Na^+/H^+ exchanger type 1 (NHE-1), may reduce intracellular Na^+ and secondarily limit Ca^2+ overload. Early clinical findings revealed improved biomarkers and tolerable safety but require larger trials.
Oxidative Stress
Excess cytosolic Ca^2+ fosters overproduction of reactive oxygen species (ROS), damaging muscle proteins, lipids, and mitochondria .
-
- Coenzyme Q10 (CoQ10) helps preserve mitochondrial function and scavenge free radicals. Preclinical data in mdx mice demonstrated a ∼42% improvement in muscle strength. A Phase 3 trial found potential respiratory benefits with acceptable safety.
-
- Idebenone, a synthetic analog of CoQ10, has shown reduced oxidative stress, improved cardiac function, and better exercise tolerance in mdx mice. Despite positive data on respiratory function, improvements in upper limb strength remain modest in clinical trials (DELOS, SYROS).
-
- N-acetylcysteine (NAC), a potent antioxidant, can decrease inflammation and necrosis in mdx mice, though it may also suppress muscle mass gain. Translational studies must balance antioxidant benefits against potential interference with muscle growth.
Muscle Ischemia
Dystrophin deficiency reduces sarcolemmal localization of neuronal nitric oxide synthase (nNOS), impairs muscle vasodilation and oxygen delivery.
-
- Phosphodiesterase type 5 (PDE5) inhibitors like Tadalafil and Sildenafil enhance the NO-cGMP signaling, potentially mitigating muscle ischemia. Tadalafil showed promising preclinical results but failed to demonstrate efficacy in Phase 3 trials for functional endpoints in ambulatory DMD. Sildenafil similarly showed no significant functional benefit.
-
- L-arginine supplementation can restore NO production. Combined L-arginine–metformin therapy has been reported to slow muscle decline in some DMD trials, though further data are needed.
Muscle Atrophy
Muscle mass maintenance depends on balancing protein synthesis and degradation.
-
- β2-Adrenergic agonists (e.g., Clenbuterol, Formoterol) can stimulate myofiber hypertrophy and satellite cell proliferation. DT-200, designed for sarcopenia, improved running performance and muscle mass in mdx mice. Human trials in DMD remain pending.
-
- Urocortins (Ucns), natural agonists of corticotropin-releasing factor receptors, can slow muscle degeneration by activating cAMP-related pathways. Animal studies suggest improvements in diaphragm function, but human data are not yet available.
-
- Myostatin inhibition is a compelling approach, as myostatin negatively regulates muscle growth. Inactivating myostatin in mdx mice increases muscle mass, with strategies including neutralizing antibodies or Follistatin (FST) gene delivery. AAV1.CMV.huFollistatin344 is under Phase 1/2 investigation in DMD (NCT02354781). Balancing muscle hypertrophy with cardiovascular safety will be critical.
Bone Loss
DMD patients often develop osteoporosis due to chronic glucocorticoid use, disuse atrophy, and metabolic derangements. Bisphosphonates (e.g., Pamidronate, Zoledronic acid, Alendronate) have shown benefits in improving bone density and reducing fracture risk in small-scale DMD studies. A comprehensive management plan with adequate vitamin D and calcium is essential to reduce bone fragility.
Candidate Gene Targets Predicted by Meta-Analysis
Because DMD encompasses numerous secondary pathological processes, identifying robust molecular targets is paramount.
The protein-protein interaction (PPI) network construction highlighted several hub genes based on Maximal Clique Centrality (MCC) scores. Enrichment analyses (Gene Ontology [GO], KEGG) of the robust DEGs revealed a consistent upregulation of genes linked to ECM organization, fibrosis, and immune response.
Ten hub genes emerged from the network:
-
- Fibronectin 1 (FN1)
-
- Connective Tissue Growth Factor (CTGF)
-
- Secreted Phosphoprotein 1 (SPP1, also known as Osteopontin)
-
- Periostin (POSTN)
-
- Thrombospondin 2 (THBS2)
-
- Collagen Type IV Alpha 2 (COL4A2)
-
- Transforming Growth Factor Beta 1 (TGF-β1)
-
- Galectin 3 (LGALS3)
-
- Platelet and Endothelial Cell Adhesion Molecule 1 (PECAM1)
-
- C-C Motif Chemokine Ligand 2 (CCL2)
Of these, seven (FN1, CTGF, SPP1, POSTN, THBS2, COL4A2, TGF-β1) are strongly associated with ECM formation and fibrotic pathways. The consistent finding of fibrotic mediators as high-priority DEGs corroborates the crucial role of fibrosis in DMD pathology and suggests that anti-fibrotic therapies may be central to improving muscle function.
SPP1 (Osteopontin) is especially interesting as it regulates immune cell recruitment and activates TGF-β1, thus bridging inflammation and fibrosis. Meanwhile, FN1 upregulation has been proposed as a biomarker correlating with DMD progression (90).
Collectively, these findings underscore the pathological significance of excessive ECM deposition. They may also inform the design of novel combination therapies (e.g., targeting TGF-β1–CTGF–SPP1 pathways) to curb fibrosis, giving gene- or cell-based therapies a better environment to succeed.
Discussion and Future Directions
Bottlenecks of Dystrophin-Centric Therapies
Despite immense progress in gene-based strategies (exon skipping, stop codon readthrough, micro-dystrophin gene addition, CRISPR/Cas9 editing), numerous limitations remain:
-
- Mutation-Specific Approaches: Eteplirsen, Golodirsen, and Viltolarsen each only suit patients with specific mutations amenable to exon 51/53 skipping. Wider coverage multi-exon skipping is more complex to deliver effectively.
-
- Vector Constraints: AAV has limited carrying capacity, restricting full-length dystrophin transgenes. High-dose administration can provoke immune responses, liver toxicity, or neutralizing antibodies.
-
- Safety of Gene Editing: CRISPR’s off-target effects and immunogenicity remain critical concerns for large-scale trials.
-
- Distribution: Achieving uniform transduction/expression across all skeletal muscles and the heart is challenging, given the body-wide muscle mass and individual variability in vascular perfusion.
Downstream Modulation: A Viable Adjunct or Standalone?
Targeting secondary pathological mechanisms may not fundamentally cure DMD but can significantly improve quality of life and slow disease progression. These approaches apply to a broader patient population regardless of genetic mutation. In practice, a multi-modal therapy combining partial dystrophin restoration with anti-inflammatory, anti-fibrotic, and muscle growth–enhancing interventions seems the most plausible future clinical strategy.
Yet even among downstream therapies, many face obstacles:
-
- Redundant Signaling: TGF-β blockade can be partially offset by alternative fibrogenic or inflammatory signals.
-
- Toxicities: Anti-inflammatory and anti-fibrotic therapies can risk immunosuppression and hamper muscle regeneration if not precisely dosed.
-
- Lack of Functional Endpoints: Many trials record improvements in biomarkers or surrogate measures but fail to translate these changes into robust functional outcomes (e.g., 6MWT, time to stand).
Emerging Concepts and Combination Therapies
Given the complexity of DMD pathophysiology, combination strategies are increasingly considered. For instance, pairing a gene-therapy vector with an anti-fibrotic agent could improve tissue-level microenvironments, enhancing vector distribution and cell engraftment. Similarly, a CRISPR-based gene repair approach might be augmented by an antioxidant or anti-inflammatory compound to maximize muscle fiber viability during the crucial editing period.
Additional prospective combination interventions might include:
-
- Steroid-Sparing Regimens: Use of novel anti-inflammatory agents (e.g., Vamorolone) to reduce dependence on standard glucocorticoids and their side effects.
-
- Myostatin or ActRIIB Pathway Inhibition combined with micro-dystrophin therapy, aiming to simultaneously protect muscle mass and correct the dystrophin defect.
-
- Nutraceutical and Physical Therapy synergy, e.g., CoQ10 or idebenone supplementation with carefully moderated exercise, could sustain mitochondrial function, but the optimal regimen needs evidence-based refinement.
Gene Profiling and Personalized Medicine
The meta-analysis of gene expression changes in DMD underscores the pivotal contribution of ECM and fibrotic processes. Future large-scale omics studies (e.g., single-cell RNA sequencing, proteomics) may elucidate new molecules or pathways implicated in DMD progression, especially in earlier disease stages. This might allow stratification of patients by likely response to anti-fibrotic or anti-inflammatory therapies, refining personalized treatments.
Furthermore, advanced computational modeling and network pharmacology can integrate multiple omics layers—genomics, transcriptomics, epigenomics, proteomics, metabolomics—to predict synergy between existing drugs or to identify novel repurposing opportunities.
Conclusion
Duchenne muscular dystrophy remains a formidable therapeutic challenge, with no single therapy offering a complete cure. Nonetheless, the field has progressed remarkably:
-
- Gene-based approaches (exon skipping, readthrough, gene addition, gene editing) promise to partially restore dystrophin or correct the genetic defect. Three exon-skipping drugs have received regulatory approval, although they target relatively small patient subsets and require further evidence on long-term functional gains.
-
- Cell-based therapies using myogenic stem or progenitor cells (including genetically corrected hiPSCs) show theoretical promise but have struggled with low engraftment rates and safety concerns in clinical application.
-
- Protein replacement therapies leveraging utrophin or related proteins may provide partial compensation for absent dystrophin, though results so far have been modest.
-
- Downstream interventions that target inflammation, fibrosis, oxidative stress, calcium dysregulation, muscle atrophy, and bone loss could help slow disease progression and improve quality of life. Drugs like Vamorolone (an alternative steroid), anti-fibrotic antibodies (FG-3019 targeting CTGF), and antioxidants (CoQ10, idebenone) have shown varying degrees of promise in clinical or preclinical settings.
Our meta-analysis of DMD transcriptome datasets indicates key molecular drivers revolve around extracellular matrix (e.g., TGF-β1, CTGF, FN1, POSTN), with direct implications for anti-fibrotic strategies. As research expands and clinical trials incorporate multi-pronged interventions, it is increasingly likely that successful management of DMD will combine the partial restoration of dystrophin via gene (sse lead story or cell therapy with robust targeting of fibrotic, inflammatory, and degenerative pathways.
Despite these advances, many critical questions remain:
-
- Long-term Safety: Especially for gene editing or high-dose AAV-based therapies.
-
- Efficacy Across All Muscles: Including cardiac and respiratory muscle, which dictate morbidity.
-
- Personalized Medicine: Tailoring therapies based on the patient’s mutation, disease stage, and molecular profile.
The future likely holds integrated therapeutic regimens, possibly guided by precision medicine and advanced biomarker monitoring to optimize safety and maximize functional gains. As we refine understanding of DMD’s pathogenesis and the interplay between primary dystrophin defects and secondary pathological cascades, we inch closer to meaningful, life-extending, and quality-of-life-improving solutions for patients with Duchenne muscular dystrophy.
Author’s Opinion
A synergistic approach appears most promising from the breadth of current evidence and the promising preclinical/clinical trials. While gene-editing therapies represent a potential “one-time treatment,” practical limitations regarding broad muscle targeting, immunogenicity, and off-target edits underscore the need to continue advancing parallel downstream strategies. Fibrosis, in particular, emerges as a critical and underappreciated contributor to disease burden; effective anti-fibrotic interventions may dramatically enhance the efficacy of gene-based therapies or cell transplantation protocols.
Given the rapid pace of innovation, I speculate that within the next decade, clinical regimens for DMD will likely standardize on a combination approach—e.g., a partial dystrophin-restoring therapy (via exon skipping or micro-dystrophin vector) combined with at least one anti-fibrotic agent, an optimized steroid or steroid alternative, and supportive therapies (antioxidants, gene modifiers). This multi-modal regimen may finally produce clinically meaningful, sustained improvements in muscle function, cardio-respiratory capacity, and survival.
mmmm
Final Remarks on Meta-Analysis and Potential Target Validation
Our meta-analysis clearly shows that transcripts related to inflammatory and fibrotic pathways are already dysregulated even at relatively early stages of the disease. This suggests that therapies aimed at halting or reversing the vicious cycles of ECM remodeling and immune cell infiltration might be beneficial from the earliest clinically feasible point. Indeed, the identification of a network of ECM- and fibrosis-related genes reinforces the premise that anti-fibrotic strategies may be pivotal to any future standard-of-care combination regimen for DMD.
However, several caveats should be considered: (1) The included microarray datasets vary in patient age and muscle biopsy sites, potentially masking stage-specific gene expression changes. (2) Some samples were small, possibly underpowered, emphasizing the need for integrative omics analysis in larger cohorts. (3) Tissue-level transcript changes do not always correspond directly to functional protein levels in diseased muscle. Thus, experimental follow-up is necessary to validate these targets and evaluate their roles in DMD.
Conclusion and Outlook
In summary, the multifaceted nature of Duchenne muscular dystrophy necessitates an equally multifaceted therapeutic strategy. Although genetic interventions remain central to the search for a definitive cure, the pragmatic near-term approach is likely an integrative regimen combining dystrophin-restoring methods with therapies that mitigate chronic inflammation, ECM remodeling, oxidative damage, and other downstream consequences. Our computational findings, supported by a robust meta-analysis of DMD transcriptomes, highlight fibrotic and immune pathways as core elements of disease pathology. New and ongoing clinical trials leveraging refined AAV vectors, CRISPR-based editing, advanced cell therapies, and novel anti-fibrotic compounds offer hope that more effective, personalized, and safer interventions will emerge in the coming years.
This review synthesizes multiple peer-reviewed sources, clinical trial data, and a meta-analysis of gene expression profiles. However, ongoing studies may yield new insights, and the field is rapidly evolving.